Modeling Duchenne Muscular Dystrophy in C. elegans
DMD is a lethal form of muscular dystrophy
DMD is a recessive X-linked form of muscular dystrophy, which affects approximately 1 in 3,600 males, resulting in muscle degeneration and death. DMD is caused by mutations in the dystrophin gene, a muscle protein that links the internal cytoskeletal system of individual myofibers to structural proteins within the extracellular matrix. The disease leads to progressive symmetric muscle weakness, muscle degeneration and its substitution with fat and connective tissue, causing random and progressive loss of contractile function. As of today, there is no cure for this disease. The first disease symptoms usually occur between the ages of 2 and 5, with definitive diagnosis between 5 and 6 years of age. Life expectancy is into the late 20s and 30s. Current research in DMD is aimed at restoring the production of dystrophin.
After decades of intense research, it is now clear that there are many cellular mechanisms involved in the disease process. The absence of the dystrophin gene drives transcriptional dysregulation in muscle cells. In addition, it induces chronic inflammation in the damaged muscle that both exacerbates necrosis through the production of free radicals and promotes the production of large fibrotic scar tissue that limits muscle function. Ultimately, DMD affects many body systems including the musculoskeletal, cardiovascular, respiratory, and immune system that reduces the lifespan of the individual.
The dystrophin glycoprotein complex (DGC) has both structural and signaling roles
The dystrophin gene codes for a structural protein found beneath the sarcolemma, where it is anchored both to the dystrophin glycoprotein complex (DGC) and cytoskeletal actin, thus stabilizing the protein complex and the integrity of the cell membrane.
The Location of the Dystrophin Complex (DGC).
Many components of the DGC have been identified so far, including the dystrobrevins (DB-α/β), the syntrophins (α1, β1, β2, γ1, and γ2), dystroglycan (DG-α/β), the sarcoglycans (SG-α, SG-β, SG-γ, SG-δ, SG-ε, and SG-ζ), and sarcospan. Dystrophin binds to the DGC using a cysteine-rich region containing a WW- and a ZZ- domains present at its C-terminal end, and to the cytoskeleton F-actin using two calponin homology (CH) domains located at its N-terminal end.
While the structural role of dystrophin and the DGC is well characterized, we still do not fully understand the signaling role executed by the dystrophin gene itself and in the relationship with the DGC. For example, dystrophin was recently found targeted by the signaling protein calmodulin, a messenger protein activated by Ca2+ molecules and controls downstream regulatory proteins such as kinases and phosphatases. The reason for this interaction is not clear, but calmodulin is known to mediate many crucial cellular processes, such as inflammation, metabolism, apoptosis, and smooth muscle contraction. In vitro assay studies using deletion mutants showed that the Ca2+-calmodulin dependent kinase phosphorylates the C-terminal domain of dystrophin, and in a shorter neuronal isoform of dystrophin this phosphorylation modulates its re-localization to the nucleus in cell lines, suggesting an additional signaling function. Dystrophin's signaling activity also cross-talks with other signaling factors including cation and water channels (5), kinases and nNOSμ, which synthesizes the majority of nitric oxide (NO) in mature skeletal muscle cells. nNOSμ function only when is localized to the sarcolemma by binding both the dystrophin gene and its binding partner α-syntrophin, where overrides vasoconstriction and prevents functional ischemia in contracting muscles. nNOSμ function is severely impaired in the dystrophin-deficient skeletal muscles of mdx mice and individuals with DMD.
A novel model to study Duchenne Muscular Dystrophy
While there are several in vivo and in vitro models to study DMD, it is still unclear how the muscle transcriptome rearranges overtime in the absence of the dystrophin gene. While the usage of the mdx mouse model increased our understanding of how this disease manifests itself and progresses, the community could benefit from alternative DMD models to complement mdx mice research accelerating translational research.
Recently, a new and very promising C. elegans DMD model became available, allowing for the study of this disease in a simple tractable organism. This strain lacks the worm homology of the dystrophin gene and presents many features of the disease such as a shortened lifespan and progressive muscle paralysis, which can be rescued by reintroducing the human dystrophin gene. We have performed genetic crosses to establish transgenic C. elegans strains that serve both as a model for DMD, and as a functional tool to perform high quality, muscle-specific RNA-IPs at a resolution that has not yet been achieved. We have sequenced and analyzed muscle specific transcriptomes at two distinct developmental stages during disease progression and identified several differentially regulated pathways in dystrophic nematode muscle. The striking similarity in disrupted pathways between worms, the mdx mice model, and DMD patients suggests that although with some differences, the genetic players and their involvement in this disease are conserved throughout evolution. In addition, C. elegans allows experimentation, which cannot be performed with current models, and could provide important new insights into this disease.
Our lab studies and detects and map high-quality muscle-specific variations in gene expression changes due to loss of the dystrophin gene at different stages of the disease. Our goal is to gain mechanistic insights of the worm DGC complex in normal states and absence of functional dystrophin gene.
Genome-wide RNAi screen for genetic interactors of the dystrophin gene in C. elegans.
We have developed a novel, temperature-based genetic screen and performed a quantitative analysis to assess the functional significance of the identified genes in the viability of muscle cells, thus confirming their contribution towards DMD initiation and progression.
A fully automated pipeline to detect genetic interactors of the dystrophin gene. The system is produced by HighRes Biosolutions and consists in a collection of freezers, coolers, liquid handler robots, plate readers, labellers, peelers, incubators and centrifuges disposed in a circle and connected by three robotic arms that move the experimental plates in a carousel fashion between the instruments.
This novel synthetic lethality assay allows a direct evaluation of the role of specific genetic pathways in the clinical severity of DMD, which may be fruitful in developing drug targets for treating DMD patients. Our preliminary data shows that it can rapidly identify novel dys-1 interactors that enhance paralysis in dys-1 dependent manner. These genes could be novel members of the worm DGC, signaling factors that directly or indirectly communicate with dys-1 and/or the DGC, and genes broadly involved in ameliorating the paralysis.
Our screening method is now mature for high throughput, genome-wide RNAi screens, as we have adapted it to 48-well microplates in liquid screen. This screen can provide a platform for rapidly identifying potential therapeutic targets that can be first validated in mammalian model systems, and eventually considered as part of a treatment plan for human DMD patients.
GFP Images = C. elegans strain lacking functional dystrophin gene (dys-1).
Credit Dr. Jason Newbern

...A worm to study Duchenne Muscular Dystrophy…C. elegans strain lacking the dystrophin gene 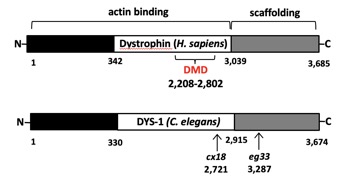 Human Dystrophin gene vs the C. elegans ortholog dys-1 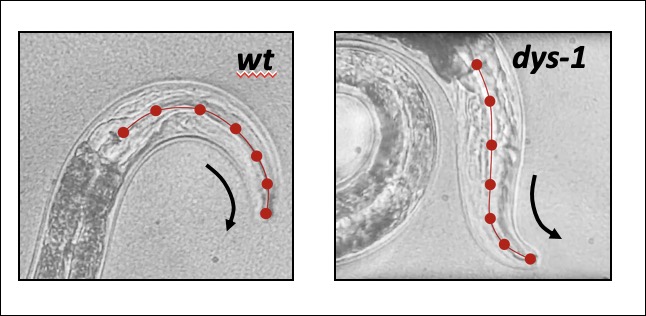 The Dystrophin-deficient worm strains dys-1(cx18) and dys-1(eg33) exhibit a head bending phenotype. wt head bending coincides with direction of movement (black arrows), while dys-1 head bending opposes direction of movement. 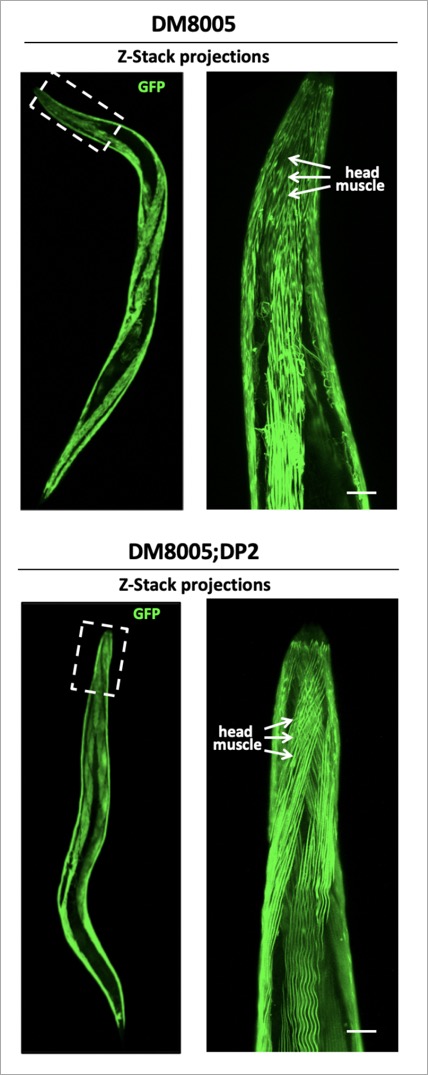 Confocal images of wt (DM8005, [myo-3p::GFP::myo-3 + rol-6(su1006)]) and dystrophin deficient strains ([myo-3p::GFP::myo-3 + rol-6(su1006)];DP1) for myosin heavy chain fibres. Right panels show an enlargement of the head muscles in both strains. 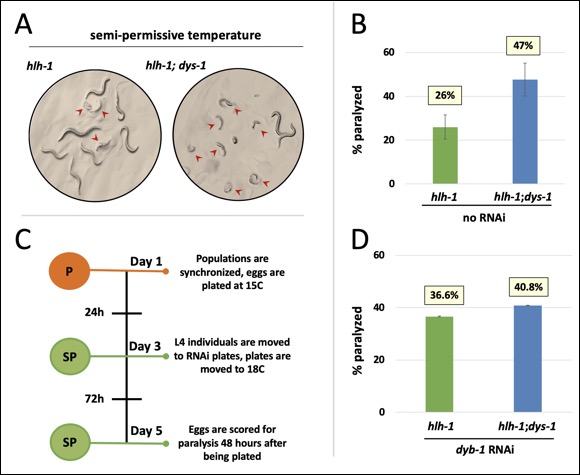 A innovative genome-wide synthetic lethality screen in C. elegans to detect dystrophin-linked genes in the nematode C. elegans. The screen is simple and effective. The RNAi response will be induced through feeding in hlh-1;dys-1(cx18) and dys-1(cx18) worm strains. The level of paralysis will be assessed using the CellProfiler worm toolbox software by comparing the degree on paralysis observed between these two strains at the semi-permissive temperature of 18°C. |